Research
Our research crosses a broad spectrum of areas, centered around microbial evolutionary genomics and antibiotic resistance. Our projects are curiosity-driven and range from fundamental evolutionary questions, to genetic arms races, to algorithmic and technology development. We work at the intersection of experiment, computation, theory, and engineering, focusing on how best to answer the scientific question and bringing to bear whatever tools necessary, even if it means building new ones.
Mobile genetic element evolution
Perhaps the most profound way that bacterial evolution differs from that of metazoa is frequent horizontal gene transfer. Horizontal gene transfer, the sharing of fragments of genetic material, allow bacteria to shortcut what would otherwise be a long evolutionary process to gain new function. Even more interestingly, horizontal transfer is often not controlled by the bacteria themselves but mediated by mobile genetic elements, acellular replicating units including plasmids, integrative conjugative elements, bacteriophages, and the murky things that don’t fit cleanly into a category.
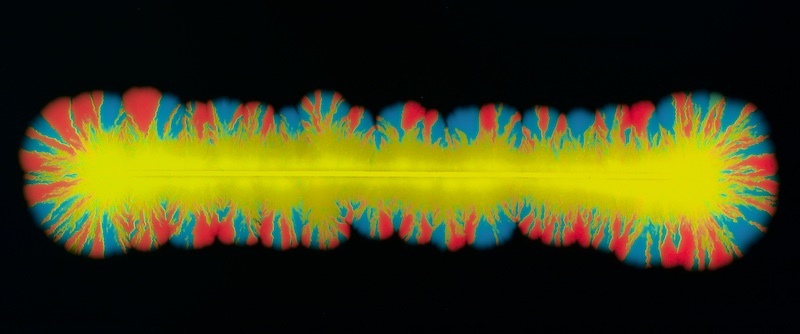
We study how mobile genetic elements evolve, and how that evolution influences and conflicts with their bacterial hosts. We primarily study plasmids, though transposons, phage, and integrative-conjugative elements, and more exotic MGEs are certainly of interest as well.
Bacteriophage discovery and biology
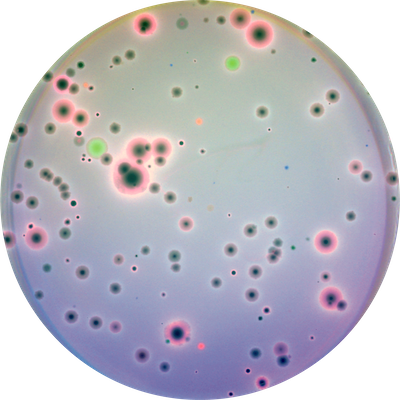
Bacteriophages, viruses that infect bacteria, are the most numerous and diverse biological entity on the planet. We develop techniques for discovering new bacteriophages, and study their evolution and interactions with their hosts, other mobile genetic elements, and each other.
Synthetic tools for microbial genomics
Precision study of microbial genetics requires the development of new tools. We engineer new fluorophores, strains, and molecular barcoding systems to precisely manipulate and measure evolving microbial systems. When the best way to do something isn't something that already exists, we build it.

Evolutionary Tradeoffs in Antibiotic Resistance
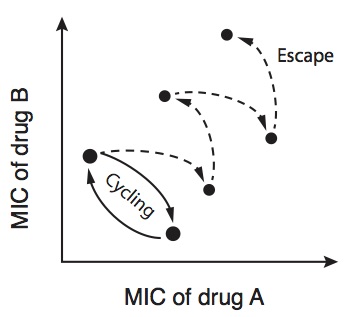
In the laboratory, antibiotic resistance evolves and fixes readily, but on closer inspection this widely-held model of resistance evolution fails to explain several critical features seen in the wild. This implies the existing model of resistance evolution is incomplete, and in particular that there exist evolutionary factors which have a countervailing effect on resistance evolution of comparable magnitude to the human use of antibiotics. We study these tradeoffs with the hope that it might inform new, evolutionarily-guided approaches to controlling the spread of antibiotic resistance.
Algorithms for Microbial Genomics
Modern genomics has transformed how we study microbial systems. With long read and real-time sequencing technologies, we are facing another leap in how we understand the molecular genetics of microorganisms. In order to make sense of this data, we need new algorithms that are both robust and scale well. We develop approaches that use a combination of graph algorithms and insights from the structure of the data to extract new understanding of bacterial function and evolution.
